New genes
born by accident
lead to evolutionary innovation
Evolution, Genetics
Novel genes are continuously emerging during evolution, but what drives this process?
A new study, published in PLOS Genetics, has found that the fortuitous appearance of certain combinations of elements in the genome can lead to the generation of new genes.
This work was led by Jorge Ruiz-Orera and Mar Alba from Hospital del Mar Medical Research Institute in Barcelona (IMIM-ICREA).
A new study has found that the fortuitous appearance of certain combinations of elements in the genome can lead to the generation of new genes [Credit: National Human Genome Research Institute]
In every genome, there are sets of genes, which are unique to that particular species.
In this study, the scientists first identified thousands of genes that were specific to human or chimpanzee.
Then, they searched the macaque genome and discovered that this species had significantly less element motifs in the corresponding genomic sequences.
These motifs are recognized by proteins that activate gene expression, a necessary step in the formation of a new gene. The formation of genes de novo from previously non-active parts of the genome was, until recently, considered highly improbable.
This study has shown that the mutations that occur normally in our genetic material may be sufficient to explain how this happens.
Once expressed, the genes can act as a substrate for the evolution of new molecular functions.
This study identified several candidate human proteins that bear no resemblance to any other known protein.
What they do is an enigma still to be resolved.
Source: PLOS [December 31, 2015]
Visualizzazione post con etichetta evoluzione. Mostra tutti i post
Visualizzazione post con etichetta evoluzione. Mostra tutti i post
martedì 27 ottobre 2015
Geni Mobili ed Evoluzione.
Scientists discover protein factories
hidden in
human jumping genes
Breakingnews, Evolution, Genetics, Human Evolution
Scientists have discovered a previously unknown wellspring of genetic diversity in humans, chimps and most other primates.
This diversity arises from a new component of itinerant sections of genetic code known as jumping genes.
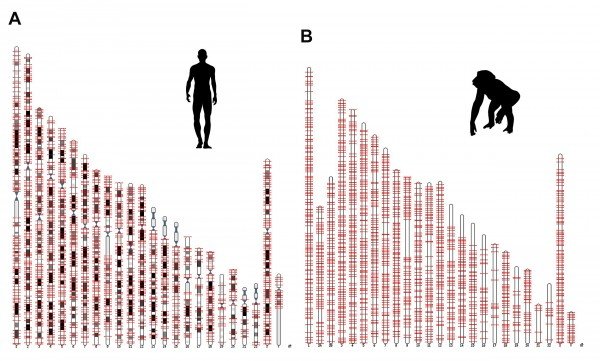
In a paper published October 22, 2015 in Cell, Salk scientists report finding human and chimp DNA peppered with sequences of genetic code they've dubbed ORF0, which spreads throughout the genome on jumping genes.
The ORF0 sequences may produce hundreds or even thousands of previously unknown proteins.
The abundance of ORF0 instances in the human genome suggests that it played--and still plays--an important role in evolutionary diversity and flexibility by serving as a mechanism for generating novel proteins. The discovery of these mobile protein factories may also shine light on the origins of genetic mutations responsible for cancer, mental disorders and other diseases.
"This discovery shows that jumping genes are an even more important source of variation in the primate genome than we thought, whether you're looking at the level of different species, different people or even the different cells within an individual's body," says Rusty Gage, senior author of the paper and a professor in Salk's Laboratory of Genetics.
With the sequencing of the human genome, it became clear that jumping genes--mobile genetic elements first discovered in maize by Barbara McClintock in the early 1950s--were also present and highly active during human evolution.
About half of the human genome resulted from sequences of genetic code that moved or insert extra copies of themselves throughout the genome.
The evolutionary importance of jumping genes was highlighted by the results of another recent study by Gage and collaborators at Stanford. The research used stem cell technologies developed in Gage's lab to explore how differences in gene expression contribute to human and chimp facial structure.
The findings, also reported in Cell, suggested that jumping genes played a role in the evolutionary split between humans and other primates.
In the more recent study that uncovered ORF0, Gage and his colleagues focused on a class of jumping genes known as LINE-1 elements, which make up about 17 percent of the human genome.
These elements contain all the necessary genetic machinery for moving themselves and other classes of jumping genes, unaided, to elsewhere in the genome.
Previously, it was thought that LINE-1 elements contain just two sequences that coded for proteins, the ultimate product of genes that serve a wide range of roles in our cells and organs.
These sequences are known as open reading frames (ORF), and the two previously known sequences, ORF1 and ORF2, are thought to be involved in producing proteins that are important for allowing LINE-1 elements to move around in the genome. In their new study, Gage and his colleagues discovered a third open reading frame. They named it ORF0 based on its location in the LINE-1 element next to ORF1. The scientists found ORF0 in about 3,500 locations in the DNA of humans and about 3,000 locations in the chimp and most other primate genomes.
The structure of LINE-1 elements is such that when an element moves to another site there is a possibility that the ORF0 sequence can blend with genetic sequences in new location in DNA. The result of the new gene sequences can be a new protein. Evolutionarily speaking, this represents a way to generate entirely new molecules that could be beneficial to a species. On the other hand, the reshuffling of an existing ORF0 sequence during a jump could result in a disease-causing mutation.
"This discovery redraws the blueprint of an important piece of genetic machinery in primates, adding a completely new gear," says Ahmet Denli, a staff scientist in Gage's lab and the first author on the paper reporting the findings.
"Jumping genes with ORF0 are basically protein factories with wheels, and over the eons evolution has been driving the bus."
Now that they have identified ORF0 in the primate genome, Denli says they plan to determine how many of the instances of ORF0 actually code for proteins and to investigate what function those proteins serve. The researchers also plan to investigate the behavior of ORF0 in different cell types and diseases. They are particularly interested in exploring its role in cancers and in neurological disorders such as schizophrenia, where previous studies have suggested jumping genes may be involved.
Source: Salk Institute [October 22,2015]

hidden in
human jumping genes
Breakingnews, Evolution, Genetics, Human Evolution
Scientists have discovered a previously unknown wellspring of genetic diversity in humans, chimps and most other primates.
This diversity arises from a new component of itinerant sections of genetic code known as jumping genes.
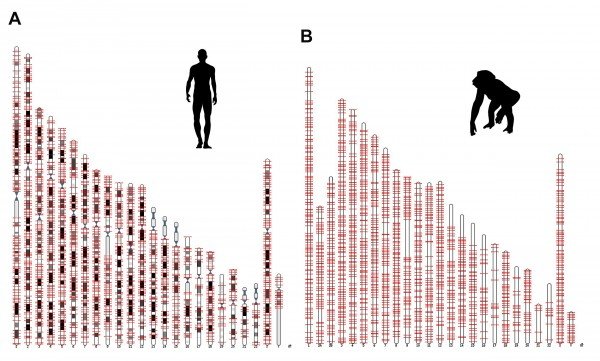
Salk researchers discovered a new genetic component, called ORF0, spread throughout the DNA of humans, chimps and most other primates. This image shows the locations of the ORFO on human and chimp chromosomes
[Credit: Salk Institute]
[Credit: Salk Institute]
In a paper published October 22, 2015 in Cell, Salk scientists report finding human and chimp DNA peppered with sequences of genetic code they've dubbed ORF0, which spreads throughout the genome on jumping genes.
The ORF0 sequences may produce hundreds or even thousands of previously unknown proteins.
The abundance of ORF0 instances in the human genome suggests that it played--and still plays--an important role in evolutionary diversity and flexibility by serving as a mechanism for generating novel proteins. The discovery of these mobile protein factories may also shine light on the origins of genetic mutations responsible for cancer, mental disorders and other diseases.
"This discovery shows that jumping genes are an even more important source of variation in the primate genome than we thought, whether you're looking at the level of different species, different people or even the different cells within an individual's body," says Rusty Gage, senior author of the paper and a professor in Salk's Laboratory of Genetics.
With the sequencing of the human genome, it became clear that jumping genes--mobile genetic elements first discovered in maize by Barbara McClintock in the early 1950s--were also present and highly active during human evolution.
About half of the human genome resulted from sequences of genetic code that moved or insert extra copies of themselves throughout the genome.
The evolutionary importance of jumping genes was highlighted by the results of another recent study by Gage and collaborators at Stanford. The research used stem cell technologies developed in Gage's lab to explore how differences in gene expression contribute to human and chimp facial structure.
The findings, also reported in Cell, suggested that jumping genes played a role in the evolutionary split between humans and other primates.
In the more recent study that uncovered ORF0, Gage and his colleagues focused on a class of jumping genes known as LINE-1 elements, which make up about 17 percent of the human genome.
These elements contain all the necessary genetic machinery for moving themselves and other classes of jumping genes, unaided, to elsewhere in the genome.
Previously, it was thought that LINE-1 elements contain just two sequences that coded for proteins, the ultimate product of genes that serve a wide range of roles in our cells and organs.
These sequences are known as open reading frames (ORF), and the two previously known sequences, ORF1 and ORF2, are thought to be involved in producing proteins that are important for allowing LINE-1 elements to move around in the genome. In their new study, Gage and his colleagues discovered a third open reading frame. They named it ORF0 based on its location in the LINE-1 element next to ORF1. The scientists found ORF0 in about 3,500 locations in the DNA of humans and about 3,000 locations in the chimp and most other primate genomes.
The structure of LINE-1 elements is such that when an element moves to another site there is a possibility that the ORF0 sequence can blend with genetic sequences in new location in DNA. The result of the new gene sequences can be a new protein. Evolutionarily speaking, this represents a way to generate entirely new molecules that could be beneficial to a species. On the other hand, the reshuffling of an existing ORF0 sequence during a jump could result in a disease-causing mutation.
"This discovery redraws the blueprint of an important piece of genetic machinery in primates, adding a completely new gear," says Ahmet Denli, a staff scientist in Gage's lab and the first author on the paper reporting the findings.
"Jumping genes with ORF0 are basically protein factories with wheels, and over the eons evolution has been driving the bus."
Now that they have identified ORF0 in the primate genome, Denli says they plan to determine how many of the instances of ORF0 actually code for proteins and to investigate what function those proteins serve. The researchers also plan to investigate the behavior of ORF0 in different cell types and diseases. They are particularly interested in exploring its role in cancers and in neurological disorders such as schizophrenia, where previous studies have suggested jumping genes may be involved.
Source: Salk Institute [October 22,2015]

domenica 9 agosto 2015
L'aiuto della Genetica

Using modern human genetics to study ancient phenomena
Anthropology, Breakingnews, Genetics, Human Evolution
We humans are obsessed with determining our origins, hoping to reveal a little of “who we are” in the process.
It is relatively simple to trace one’s genealogy back a few generations, and there are many companies and products offering such services.
But what if we wanted to trace our origins further on an evolutionary timescale and study human evolution itself? In this case, there are no written records and censuses. Instead, the study of human evolution has so far relied heavily on fossil specimens and archaeological finds.
Now, genetic tools and approaches are frequently used to answer evolutionary questions and reveal patterns of divergence that reflect different selective pressures and geographical movement.
This is particularly true for studies of human migrations out of Africa, global population divergence, and its consequences for human health.

Figure 1. Diagrammatic representation of the serial founder effect model
[Credit: Emma Whittington]
Humans Originated in Africa
The current best hypothesis suggests anatomically modern humans (AMH) arose in East Africa approximately 200,000 YBP (Years Before Present).
AMH migrated from Africa around 100,000-60,000 years ago in a series of dispersals that expanded into Europe and Asia between 60,000 and 40,000 YBP.
It has been scientifically proven that East Africa is the origin of humans, and supported by both archaeological and genetic data.
Genetic diversity is greatest in East Africa and decreases in a step-wise fashion from the equator in a pattern reflecting sequential founder populations and bottlenecks.
Figure 1 shows three populations with decreasing genetic diversity (represented by the colored circles) from left to right. The first population, with the greatest genetic diversity, represents Africa. A second population is shown migrating away from ‘Africa’ taking with it a sample of the existing genetic diversity. This forms the founding population for the next series of migrations. Each time a population migrates it represents only a sample of genetic variation existing in its founding population, and in doing so, sequential migration (such as those in Figure 1) leads to a reduction in genetic diversity with increasing distance from the first population.
Leaving Africa – Where do we go from here?
Although the location of human origin is generally accepted, there is a lack of consensus around the migration routes by which AMH left Africa and expanded globally.
There are many studies using genetic tools to identify likely migration routes, one of which is a recent PLOS One article by Veerappa et al (2015).
In this study, researchers characterized the global distribution of copy number variation, which is the variation in the number of copies of a particular gene, by high resolution genotyping of 1,115 individuals from 12 geographic populations, identifying 44,109 copy number variants (CNVs).
The CNVs carried by an individual determined their CNV genotype and by comparing CNV genotypes between all individuals from all populations, the authors determined similarity and genetic distance between populations.
The phylogenetic relationship between populations proposed a global migration map (Figure 2), in which an initial migration from the place of origin, Africa, formed a second settlement in East Asia, which is similar to a founding population seen in Figure 1.
At least five further branching events took place in the second settlement, forming populations globally. The migration routes identified in this paper largely support those already proposed, but of particular interest this paper also proposes a novel migration route from Australia, across the Pacific, and towards the New World (shown in blue in Figure 2).
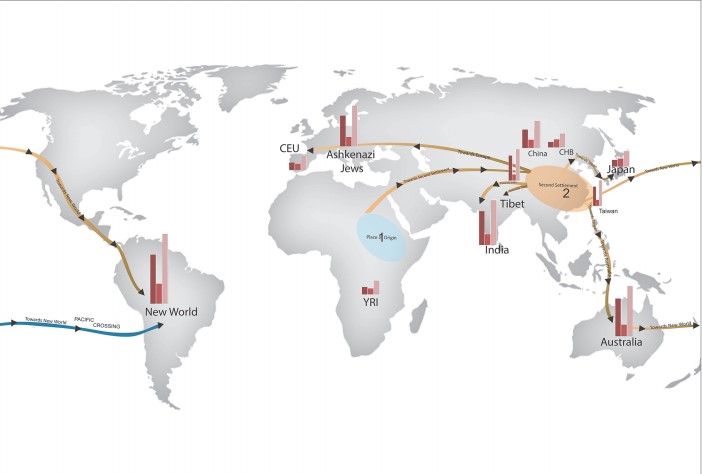 Figure 2. A global map showing CNV counts and possible migration routes
Figure 2. A global map showing CNV counts and possible migration routes
[Credit: Veerappa et al (2015)]
Global Migration Leads to Global Variation As AMH spread across the globe, populations diverged and encountered novel selective pressures to which they had to adapt. This is reflected in the phenotypic (or observable) variation seen between geographically distant populations.
At the genotype level, a number of these traits show evidence of positive selection, meaning they likely conferred some advantage in particular environments and were consequently favored by natural selection and increased in frequency.
A well cited example of this is global variation in skin color, which is thought to reflect a balance between vitamin D synthesis and photoprotection (Figure 3).
Vitamin D synthesis requires UV radiation, and a deficiency in vitamin D can result in rickets, osteoporosis, pelvic abnormalities, and a higher incidence of other diseases.
At higher latitudes, where UV radiation is low or seasonal, experiencing enough UV radiation for sufficient vitamin D synthesis is a major concern.
Presumably, as AMHs migrated from Africa, they experienced reduced levels of UV radiation, insufficient vitamin D synthesis, and severe health problems; resulting in selection for increased vitamin D synthesis and lighter skin pigmentation.
Consistent with this, a number of pigmentation genes underlying variation in skin color show evidence of positive selection in European and Asian populations, relative to Africa.
On the flip side, populations near the equator experience no shortage of UV radiation and thus synthesize sufficient vitamin D; however the risk of UV damage is much greater. Melanin, the molecule determining skin pigmentation, acts as a photoprotective filter, reducing light penetration and damage caused by UV radiation, resulting in greater photoprotection in darkly pigmented skin.
Selective pressure to maintain dark pigmentation in regions with high UV radiation is evident by the lack of genetic variation in pigment genes in areas such as Africa.
This suggests selection has acted to remove mutation and maintain the function of these genes.

Figure 3. A map showing predominant skin pigmentation globally
[Credit: Barsch (2003)]
Can genetics and human evolution have a practical use in human health?
Beyond phenotypic consequences, genetic variation between populations has a profound impact on human health.
It has a direct influence on an individual’s predisposition for certain conditions or diseases. For example, Type 2 diabetes is more prevalent in African Americans than Americans of European descent.
Genome wide association studies (GWAS) analyze common genetic variants in different individuals and assess whether particular variants are more often associated with certain traits or diseases.
Comparing the distribution and number of disease-associated variants between populations can assess if genetic risk factors underlie disparities in disease susceptibility. In the case of Type 2 diabetes, African Americans carry a greater number of risk variants than Americans of European descent at genetic locations (loci) associated with Type 2 diabetes.
It is clear that an individual’s ethnicity affects their susceptibility and likely reaction to disease, and as such should be considered in human health policy.
Understanding the genetic risk factors linking populations and disease can identify groups of individual at greater risk of developing certain diseases for the sake of prioritizing treatment and prevention.
Applying modern human genetics to human evolution has opened a door to studying ancient evolutionary phenomena and patterns.
This area not only serves to quench the desire to understand our origins, but profoundly impacts human health in a way that could revolutionize disease treatment and prevention.
In this blog, I have given a brief overview of how using genetic approaches can tell us a great deal about human origins, migration and variation between populations.
In addition, I have outlined the complex genetic underpinnings behind ethnicity and disease susceptibility, which suggests an important role for population genetics in human health policy.
This blog post covers only a fraction of the vast amount of ongoing work in this field, and the often ground breaking findings.
It is unclear exactly how far genetics will take us in understanding human evolution, but the end is far from near. The potential for genetics in this field and broader feels limitless, and I for one am excited by the prospect.
 Author: Emma Whittington | Source: PLOS Blogs [August 01, 2015]
Author: Emma Whittington | Source: PLOS Blogs [August 01, 2015]
domenica 12 luglio 2015
Clima ed Evoluzione
Si sa quanto siano stretti i rapporti tra clima ed evoluzione
dell'Uomo e della Vita in genere. Gli studi (interdisciplinari)
producono sempre nuove prove scientifiche di questo fatto.
Ecco un interessante articolo sugli effetti del vulcanismo.
Volcanic eruptions that changed human history
Ancient Environment, ArchaeoHeritage, Archaeology, Breakingnews, Earth Science, Geology, Palaeontology
It is well known that large volcanic eruptions contribute to climate variability.
However, quantifying these contributions has proven challenging due to inconsistencies in both historic atmospheric data observed in ice cores and corresponding temperature variations seen in climate proxies such as tree rings.
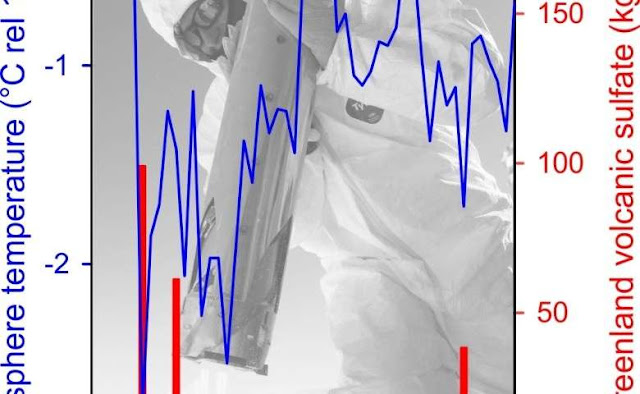
Published today in the journal Nature, a new study led by scientists from the Desert Research Institute (DRI) and collaborating international institutions, resolves these inconsistencies with a new reconstruction of the timing and associated radiative forcing of nearly 300 individual volcanic eruptions extending as far back as the early Roman period. "Using new records we are able to show that large volcanic eruptions in the tropics and high latitudes were the dominant drivers of climate variability, responsible for numerous and widespread summer cooling extremes over the past 2,500 years," said the study's lead author Michael Sigl, Ph.D., an assistant research professor at DRI and postdoctoral fellow with the Paul Scherrer Institute in Switzerland.
"These cooler temperatures were caused by large amounts of volcanic sulfate particles injected into the upper atmosphere," Sigl added, "shielding the Earth's surface from incoming solar radiation."
The study shows that 15 of the 16 coldest summers recorded between 500 BC and 1,000 AD followed large volcanic eruptions -- with four of the coldest occurring shortly after the largest volcanic events found in record.
This new reconstruction is derived from more than 20 individual ice cores extracted from ice sheets in Greenland and Antarctica and analyzed for volcanic sulfate primarily using DRI's state-of-the-art, ultra-trace chemical ice-core analytical system. These ice-core records provide a year-by-year history of atmospheric sulfate levels through time. Additional measurements including other chemical parameters were made at collaborating institutions. "We used a new method for producing the timescale," explained Mai Winstrup, Ph.D., a postdoctoral researcher at the University of Washington, Seattle. "Previously, this has been done by hand, but we used a statistical algorithm instead. Together with the state-of-the-art ice core chemistry measurements, this resulted in a more accurate dating of the ice cores." "Using a multidisciplinary approach was key to the success of this project," added Sigl.

In total, a diverse research group of 24 scientists from 18 universities and research institutes in the United States, United Kingdom, Switzerland, Germany, Denmark, and Sweden contributed to this work -- including specialists from the solar, space, climate, and geological sciences, as well as historians. The authors note that identification of new evidence found in both ice cores and corresponding tree rings allowed constraints and verification of their new age scale. "With the discovery of a distinctive signature in the ice-core records from an extra-terrestrial cosmic ray event, we had a critical time marker that we used to significantly improve the dating accuracy of the ice-core chronologies," explained Kees Welten, Ph.D., an associate research chemist from the University of California, Berkeley. A signature from this same event had been identified earlier in various tree-ring chronologies dating to 774-775 Common Era (CE). "Ice-core timescales had been misdated previously by five to ten years during the first millennium leading to inconsistencies in the proposed timing of volcanic eruptions relative to written documentary and tree-ring evidence recording the climatic responses to the same eruptions," explained Francis Ludlow, Ph.D., a postdoctoral fellow from the Yale Climate & Energy Institute. Throughout human history, sustained volcanic cooling effects on climate have triggered crop failures and famines. These events may have also contributed to pandemics and societal decline in agriculture-based communities. Together with Conor Kostick, Ph.D. from the University of Nottingham, Ludlow translated and interpreted ancient and medieval documentary records from China, Babylon (Iraq), and Europe that described unusual atmospheric observations as early as 254 years before Common Era (BCE).
These phenomena included diminished sunlight, discoloration of the solar disk, the presence of solar coronae, and deeply red twilight skies.

Tropical volcanoes and large eruptions in the Northern Hemisphere high latitudes (such as Iceland and North America) -- in 536, 626, and 939 CE, for example -- often caused severe and widespread summer cooling in the Northern Hemisphere by injecting sulfate and ash into the high atmosphere.
These particles also dimmed the atmosphere over Europe to such an extent that the effect was noted and recorded in independent archives by numerous historical eyewitnesses. Climatic impact was strongest and most persistent after clusters of two or more large eruptions.
The authors note that their findings also resolve a long-standing debate regarding the causes of one of the most severe climate crises in recent human history, starting with an 18-month "mystery cloud" or dust veil observed in the Mediterranean region beginning in March, 536, the product of a large eruption in the high-latitudes of the Northern Hemisphere.
The initial cooling was intensified when a second volcano located somewhere in the tropics erupted only four years later. In the aftermath, exceptionally cold summers were observed throughout the Northern Hemisphere.
This pattern persisted for almost fifteen years, with subsequent crop failures and famines -- likely contributing to the outbreak of the Justinian plague that spread throughout the Eastern Roman Empire from 541 to 543 CE, and which ultimately decimated the human population across Eurasia. "This new reconstruction of volcanic forcing will lead to improved climate model simulations through better quantification of the sensitivity of the climate system to volcanic influences during the past 2,500 years," noted Joe McConnell, Ph.D., a DRI research professor who developed the continuous-flow analysis system used to analyze the ice cores. "As a result," McConnell added, "climate variability observed during more recent times can be put into a multi-millennial perspective -- including time periods such as the Roman Warm Period and the times of significant cultural change such as Great Migration Period of the 6th century in Europe."
This reconciliation of ice-core records and other records of past environmental change will help define the role that large climatic perturbations may have had in the rise and fall of civilizations throughout human history. "With new high-resolution records emerging from ice cores in Greenland and Antarctica, it will be possible to extend this reconstruction of volcanic forcing probably all the way back into the last Ice Age," said Sigl.
Source: Desert Research Institute [July 08, 2015]
dell'Uomo e della Vita in genere. Gli studi (interdisciplinari)
producono sempre nuove prove scientifiche di questo fatto.
Ecco un interessante articolo sugli effetti del vulcanismo.
Volcanic eruptions that changed human history
Ancient Environment, ArchaeoHeritage, Archaeology, Breakingnews, Earth Science, Geology, Palaeontology
It is well known that large volcanic eruptions contribute to climate variability.
However, quantifying these contributions has proven challenging due to inconsistencies in both historic atmospheric data observed in ice cores and corresponding temperature variations seen in climate proxies such as tree rings.

Strong and widespread cooling occurred in the immediate aftermath of large volcanic eruptions as indicated by ice cores from Greenland [Credit: Michael Sigl]
Published today in the journal Nature, a new study led by scientists from the Desert Research Institute (DRI) and collaborating international institutions, resolves these inconsistencies with a new reconstruction of the timing and associated radiative forcing of nearly 300 individual volcanic eruptions extending as far back as the early Roman period. "Using new records we are able to show that large volcanic eruptions in the tropics and high latitudes were the dominant drivers of climate variability, responsible for numerous and widespread summer cooling extremes over the past 2,500 years," said the study's lead author Michael Sigl, Ph.D., an assistant research professor at DRI and postdoctoral fellow with the Paul Scherrer Institute in Switzerland.
"These cooler temperatures were caused by large amounts of volcanic sulfate particles injected into the upper atmosphere," Sigl added, "shielding the Earth's surface from incoming solar radiation."
The study shows that 15 of the 16 coldest summers recorded between 500 BC and 1,000 AD followed large volcanic eruptions -- with four of the coldest occurring shortly after the largest volcanic events found in record.
This new reconstruction is derived from more than 20 individual ice cores extracted from ice sheets in Greenland and Antarctica and analyzed for volcanic sulfate primarily using DRI's state-of-the-art, ultra-trace chemical ice-core analytical system. These ice-core records provide a year-by-year history of atmospheric sulfate levels through time. Additional measurements including other chemical parameters were made at collaborating institutions. "We used a new method for producing the timescale," explained Mai Winstrup, Ph.D., a postdoctoral researcher at the University of Washington, Seattle. "Previously, this has been done by hand, but we used a statistical algorithm instead. Together with the state-of-the-art ice core chemistry measurements, this resulted in a more accurate dating of the ice cores." "Using a multidisciplinary approach was key to the success of this project," added Sigl.

Narrow and distorted tree-rings from long living bristlecone-pines (Snake Mountains, Nevada, USA), indicating extreme cooling after a large volcanic eruption in 44 BCE, the year of Julius Cesar's death [Credit: Matthew Salzer]
In total, a diverse research group of 24 scientists from 18 universities and research institutes in the United States, United Kingdom, Switzerland, Germany, Denmark, and Sweden contributed to this work -- including specialists from the solar, space, climate, and geological sciences, as well as historians. The authors note that identification of new evidence found in both ice cores and corresponding tree rings allowed constraints and verification of their new age scale. "With the discovery of a distinctive signature in the ice-core records from an extra-terrestrial cosmic ray event, we had a critical time marker that we used to significantly improve the dating accuracy of the ice-core chronologies," explained Kees Welten, Ph.D., an associate research chemist from the University of California, Berkeley. A signature from this same event had been identified earlier in various tree-ring chronologies dating to 774-775 Common Era (CE). "Ice-core timescales had been misdated previously by five to ten years during the first millennium leading to inconsistencies in the proposed timing of volcanic eruptions relative to written documentary and tree-ring evidence recording the climatic responses to the same eruptions," explained Francis Ludlow, Ph.D., a postdoctoral fellow from the Yale Climate & Energy Institute. Throughout human history, sustained volcanic cooling effects on climate have triggered crop failures and famines. These events may have also contributed to pandemics and societal decline in agriculture-based communities. Together with Conor Kostick, Ph.D. from the University of Nottingham, Ludlow translated and interpreted ancient and medieval documentary records from China, Babylon (Iraq), and Europe that described unusual atmospheric observations as early as 254 years before Common Era (BCE).
These phenomena included diminished sunlight, discoloration of the solar disk, the presence of solar coronae, and deeply red twilight skies.

A freshly drilled ice-core from TUNU, Greenland containing a history of volcanic eruptions is pushed out of the core barell [Credit: Olivia Maselli]
Tropical volcanoes and large eruptions in the Northern Hemisphere high latitudes (such as Iceland and North America) -- in 536, 626, and 939 CE, for example -- often caused severe and widespread summer cooling in the Northern Hemisphere by injecting sulfate and ash into the high atmosphere.
These particles also dimmed the atmosphere over Europe to such an extent that the effect was noted and recorded in independent archives by numerous historical eyewitnesses. Climatic impact was strongest and most persistent after clusters of two or more large eruptions.
The authors note that their findings also resolve a long-standing debate regarding the causes of one of the most severe climate crises in recent human history, starting with an 18-month "mystery cloud" or dust veil observed in the Mediterranean region beginning in March, 536, the product of a large eruption in the high-latitudes of the Northern Hemisphere.
The initial cooling was intensified when a second volcano located somewhere in the tropics erupted only four years later. In the aftermath, exceptionally cold summers were observed throughout the Northern Hemisphere.
This pattern persisted for almost fifteen years, with subsequent crop failures and famines -- likely contributing to the outbreak of the Justinian plague that spread throughout the Eastern Roman Empire from 541 to 543 CE, and which ultimately decimated the human population across Eurasia. "This new reconstruction of volcanic forcing will lead to improved climate model simulations through better quantification of the sensitivity of the climate system to volcanic influences during the past 2,500 years," noted Joe McConnell, Ph.D., a DRI research professor who developed the continuous-flow analysis system used to analyze the ice cores. "As a result," McConnell added, "climate variability observed during more recent times can be put into a multi-millennial perspective -- including time periods such as the Roman Warm Period and the times of significant cultural change such as Great Migration Period of the 6th century in Europe."
This reconciliation of ice-core records and other records of past environmental change will help define the role that large climatic perturbations may have had in the rise and fall of civilizations throughout human history. "With new high-resolution records emerging from ice cores in Greenland and Antarctica, it will be possible to extend this reconstruction of volcanic forcing probably all the way back into the last Ice Age," said Sigl.
Source: Desert Research Institute [July 08, 2015]
domenica 5 luglio 2015
La Peste e la Storia
How small genetic change in Yersinia pestis changed human history
Breakingnews, Evolution, Genetics
While studying Yersinia pestis, the bacteria responsible for epidemics of plague such as the Black Death, Wyndham Lathem, Ph.D., assistant professor in microbiology-immunology at Northwestern University Feinberg School of Medicine, found a single small genetic change that fundamentally influenced the evolution of the deadly pathogen, and thus the course of human history.


In a paper published in Nature Communications, Lathem and first author Daniel Zimbler, Ph.D., a Feinberg post-doctoral fellow, demonstrated how the acquisition of a single gene caused the shift of Y. pestis from causing a primarily gastrointestinal infection to a more serious and often fatal respiratory disease.
They further showed how later modifications of this gene enhanced infections associated with the bubonic plague.
"Our findings demonstrate how Y. pestis had the ability to cause a severe respiratory disease very early in its evolution. This research helps us better understand how bacteria can adapt to new host environments to cause disease by acquiring small bits of DNA," Lathem said. The team examined ancestral strains of the bacteria in mouse models to learn when Y. pestis gained the ability to infect the lungs and cause the severe form of the disease known as pneumonic plague. In the most ancestral of all currently existing Y. pestis strains, they showed how the bacteria could successfully colonize the lungs but could not cause the severe disease associated with pneumonic plague. The biggest difference they found between this strain and closely related strains that could cause pneumonic plague was a gene for the surface protein Pla.
Lathem proposed that the bacteria's acquisition of the gene Pla enhanced its ability to cause infection in the lungs and was all that this ancestral strain of Y. pestis needed to produce a fatal lung infection.
So Lathem and his team inserted the Pla gene into this strain to observe changes in the health of the lungs. They found the newly mutated strain had gained the ability to cause respiratory infection identically to modern strains of Y. pestis that cause disease today, demonstrating that the Pla gene was necessary for Y. pestis to infect the lungs. In addition, they found that no other changes to Y. pestis were required, even though the bacteria has continued to gain and lose genes over the last several thousand years. The lab also looked at variations of the gene Pla and discovered that a single modification only found in modern strains of Y. pestis was a critical adaptation for the bacteria to spread in the body and infect the lymph nodes, a form of the infection that causes bubonic plague. According to Lathem, the surprising conclusion from this aspect of the study is that, contrary to current thinking in the field, Y. pestis may have first evolved as a respiratory pathogen before it could cause the more common form of disease, bubonic plague. Lathem said the new research may explain how Y. pestis transitioned from causing only localized outbreaks of plague to the pandemic spread of Y. pestis such as the sixth century's Justinian Plague and the fourteenth century's Black Death. "Our data suggests that the insertion and then subsequent mutation of Pla allowed for new, rapidly evolving strains of disease," Lathem said. "This information can show how new respiratory pathogens could emerge with only small genetic changes."
Author: Sarah Plumridge | Source: Northwestern University [June 30, 2015]
Breakingnews, Evolution, Genetics
While studying Yersinia pestis, the bacteria responsible for epidemics of plague such as the Black Death, Wyndham Lathem, Ph.D., assistant professor in microbiology-immunology at Northwestern University Feinberg School of Medicine, found a single small genetic change that fundamentally influenced the evolution of the deadly pathogen, and thus the course of human history.


This image of lung tissue shows inflammatory lesions in an ancestral strain of the bacteria that causes pneumonic plague without the Pla gene (above) and with the gene (below). The addition of Pla allowed this strain to cause pneumonic plague
[Credit: Northwestern University]
[Credit: Northwestern University]
In a paper published in Nature Communications, Lathem and first author Daniel Zimbler, Ph.D., a Feinberg post-doctoral fellow, demonstrated how the acquisition of a single gene caused the shift of Y. pestis from causing a primarily gastrointestinal infection to a more serious and often fatal respiratory disease.
They further showed how later modifications of this gene enhanced infections associated with the bubonic plague.
"Our findings demonstrate how Y. pestis had the ability to cause a severe respiratory disease very early in its evolution. This research helps us better understand how bacteria can adapt to new host environments to cause disease by acquiring small bits of DNA," Lathem said. The team examined ancestral strains of the bacteria in mouse models to learn when Y. pestis gained the ability to infect the lungs and cause the severe form of the disease known as pneumonic plague. In the most ancestral of all currently existing Y. pestis strains, they showed how the bacteria could successfully colonize the lungs but could not cause the severe disease associated with pneumonic plague. The biggest difference they found between this strain and closely related strains that could cause pneumonic plague was a gene for the surface protein Pla.
Lathem proposed that the bacteria's acquisition of the gene Pla enhanced its ability to cause infection in the lungs and was all that this ancestral strain of Y. pestis needed to produce a fatal lung infection.
So Lathem and his team inserted the Pla gene into this strain to observe changes in the health of the lungs. They found the newly mutated strain had gained the ability to cause respiratory infection identically to modern strains of Y. pestis that cause disease today, demonstrating that the Pla gene was necessary for Y. pestis to infect the lungs. In addition, they found that no other changes to Y. pestis were required, even though the bacteria has continued to gain and lose genes over the last several thousand years. The lab also looked at variations of the gene Pla and discovered that a single modification only found in modern strains of Y. pestis was a critical adaptation for the bacteria to spread in the body and infect the lymph nodes, a form of the infection that causes bubonic plague. According to Lathem, the surprising conclusion from this aspect of the study is that, contrary to current thinking in the field, Y. pestis may have first evolved as a respiratory pathogen before it could cause the more common form of disease, bubonic plague. Lathem said the new research may explain how Y. pestis transitioned from causing only localized outbreaks of plague to the pandemic spread of Y. pestis such as the sixth century's Justinian Plague and the fourteenth century's Black Death. "Our data suggests that the insertion and then subsequent mutation of Pla allowed for new, rapidly evolving strains of disease," Lathem said. "This information can show how new respiratory pathogens could emerge with only small genetic changes."
Author: Sarah Plumridge | Source: Northwestern University [June 30, 2015]
venerdì 15 maggio 2015
7. DNA non codificante: significato.

Il DNA non codificante è solamente un rimasuglio insignificante di materiale genetico estraneo introdotto (proditoriamente, aggressivamente, per errore) in tempi passati, in vari modi, da vari agenti estranei?
E' solamente ciò che resta di antico materiale nucleare ormai inutile ed inutilizzabile per il nostro organismo moderno?
Se possiede qualche funzione, è questa funzione solamente di 'protezione' generica del DNA codificante - l'unico inteso come prezioso - contro le mutazioni?
No. Anzi, si tratta di un argomento del quale possono parlare unicamente gli esperti (che spesso, si è visto, sbagliano anch'essi, in questo campo sconosciuto). E tutti gli altri è bene che tacciano.
Si può affermare che la ‘lezione’ impartitaci fino ad oggi dal DNA ‘ciarpame’ sia quella di
adottare un’enorme prudenza nelle future valutazioni.
Il DNA ‘ciarpame’ (e non ‘spazzatura’: è un errore di
traduzione!) è stato infatti riabilitato a DNA‘non codificante’: e persino
questa definizione più politicamente corretta è stata riconosciuta come almeno
in parte sbagliata, perché alcune porzioni del DNA non codificante codificano
eccome!
Il Codice Genetico è una lingua ancora in grandissima parte
sconosciuta.


Esempi dell’importanza del DNA ‘non codificante’.
La semplice ‘regolazione’ dell’espressione genetica possiede
enormi effetti sulla fisiologia degli esseri viventi.
Nelle porzioni di DNA non codificante (ed anche nei geni)
sono state reperite variazioni di una sola lettera che influenzano il rischio di malattia. Si è visto
che esiste una rara mutazione (nel Gene MC4R) che è responsabile dell’obesità
infantile. In soggetti con normali MC4R, si è però osservata una tendenza
ancora maggiore all’obesità: si è visto che questi soggetti avevano ereditatao
una frequente variante del DNA ‘ciarpame’ circostante al gene MC4R, con
funzioni che alterano la sua attività.
Il DNA non codificante può essere anche responsabile delle
differenze esistenti tracce diverse, seppure simili. Uomo e Scimpanzè, ad
esempio, hanno il 99% dei geni in comune, ma solo il 96% del DNA complessivo. È
comunque un’affinità estremamente alta.
Ma a cosa sono dovute – allora – le enormi differenze
osservabili, quali intelligenza e linguaggio?
Ebbene: esistono differenze molto maggiori, tra le due
specie, che sono osservabili proprio nel DNA non codificante. Probabilmente
sono proprio loro il fondamento di quelle caratteristiche squisitamente umane…
Pertanto, si può concludere che sia certamente errato
considerare come unica parte importante del genoma umano i geni codificanti le
proteine…


Nè troppo, né troppo poco.
Probabilmente è un errore altrettanto grande quello di
considerare la parte di DNA non codificante alla stregua di un campo nel quale
siano seppelliti chissà quali grandi tesori.
Nel 2000 già si sapeva che le cellule umane (Rinn et al.)
leggono migliaia di segmenti di DNA, non esclusivamente le parti codificanti, e
producono molecole di RNA. Si cominciò quindi a ricercare le possibili funzioni
di queste molecole di RNA.
Dopo circa un paio di anni di ricerche (Rinn e Chang,
Stanford University) trovarono una molecola di RNA che era prodotta dalle
cellule cutanee della metà inferiore del corpo, ma non da quelle della metà
superiore. Decisero di chiamarla “Hotair” (riferendosi ad un’espressione
inglese simile alla nostra di ‘acqua calda’, o ‘aria fritta’, col dire: “ Se
alla fine risulterà essere solamente ‘hotair’, ebbene, almeno ci abbiamo
provato”).
Hotair risultò legarsi ad una particolare proteina che fu
chiamata Polycomb, che appartiene ad un gruppo di proteine estremamente
importanti per il normale sviluppo degli animali fino a partire dall’uovo
fertilizzato. La loro funzione è quella di attivare e disattivare
alternativamente i geni, in modo che un informe ‘pallottolina’ di cellule tutte
simili possa dare origine – secondo uno schema prestabilito – a tessuti
diversi, quali, muscolo, osso, cellule nervose etc.
Polycomb si sposta in siti differenti – sotto la guida di
‘Hotair’ – dove si lega con una serie di geni, spegnendoli quando è necessario.
Rinn annunciò i suoi risultati nel 2007, sulla rivista Cell:
furono giudicati grandiosi e fondamentali. Negli anni successivi, Chang
dimostrò che alcuni topi – privati di ‘hotair’ crescevano malformati in molti
modi: hotair regolava il funzionamento di molti distretti, quindi, non
solamente la cute o le ossa.
In seguito, altri studi hanno dimostrato che alcune porzioni
di DNA non codificante sono così importanti che – se rimosse – determinano la
morte dell’individuo.
Rinn ha scoperto anche una particolare molecola di RNA (che
ha chiamato ‘firre’), che possiede una spettacolare proprietà: sembra catturare
(proprio come un cow-boy in un Rodeo) insieme tre cromosomi con una specie di
‘lasso’, tirandoli uno vicino all’altro. Da qui, ha formulato l’ipotesi che
esistano centinaia di molecole con funzioni simili, o altrettanto sorprendenti.
Il principio secondo il quale il nostro Genoma funziona
diversamente anche in relazione al suo stato di aggrovigliamento maggiore o minore
e in relazione alla sua disposizione nello spazio (e quindi esposizione di
alcuni suoi tratti a proteine altrimenti inaccessibili) era già stato enunciato
da tempo.
Tra l’altro è uno dei motivi per i quali – anche se siamo
riusciti a ricostruire alcuni genomi – non sappiamo ‘impacchettare’ il DNA in
modo corretto all’interno della cellula.
L’attività riproduttiva – nel rovistare nella
valigia/cellula – finisce poi col non trovare mai i calzini, o la cintura, o la
cravatta...


Un litigio di lunga durata.
E' dai tempi di Darwin che prosegue la disputa su che cosa esattamente guidi l'evoluzione.
Darwin si lamentò molto della ‘cattiva rappresentazione’ che
veniva fatta da altri delle sue idee fondamentali, circa le forze che
determinano la grande diversità delle varie forme di vita. Un suo grande rammarico fu che molti
gli attribuissero il pensiero che solamente la Selezione Naturale ne fosse alla
base.
Certamente, Darwin era all’oscuro di DNA e Cromosomi,
Biologia Molecolare e tutto il resto: ma era altresì convinto che molte forse
guidassero l’Evoluzione, “comprese alcune variazioni che – nella nostra
ignoranza – ci sembrano insorgere spontaneamente”.
Noi facciamo molti errori.
Probabilmente, dovremmo smettere di considerare la Vita come
una continua migrazione verso la perfezione.
Probabilmente, il DNA ‘non
ricombinante’ non è un segno di insuccesso dell’Evoluzione.
Forse – invece – è proprio la traccia rimastaci
del suo lento, nascosto, inarrestabile trionfo.
giovedì 14 maggio 2015
6 - Una sesta base?
L'interrogativo è d'obbligo: sarebbe una scoperta epocale.
Classicamente, le basi sono quattro (adenina, citosina, guanina e timina).
Altre due basi derivate da queste sono la metil citosina e la metil adenina, che sono variazioni delle prime due.
Quando si scoprirono le attività biomolecolari importanti delle metil citosina (mC) essa fu aggiunta come quinta base.
Lo studio attuale, di Manuel esteller dell'Università di Barcellona e appena pubblicato sulla rivista Cell, propone che anche la metil Adenina (mA) sia 'promossa' a base; sarebbe quindi la sesta.
Essa è presente in minime quantità in vari batteri (in cui protegge dall'immissione esterna di materiale nucleare) ed in vari vegetali ed animali inferiori (alghe, vermi, mosche) e la sua funzione sarebbe quella di modulare l'attività di vari geni e di influenzare le cellule staminali.
Tutto sta a vedere se si troverà anche nei mammiferi e - in special modo - nell'uomo.
Sixth DNA's base discovered?
DNA (deoxyribonucleic acid) is the main component of our genetic material. It is formed by combining four parts: A, C, G and T (adenine, cytosine, guanine and thymine), called bases of DNA combine in thousands of possible sequences to provide the genetic variability that enables the wealth of aspects and functions of living beings.

Two more bases: the Methyl- cytosine and Methyl-adenine
In the early 80s, to these four "classic" bases of DNA was added a fifth: the methyl-cytosine (mC) derived from cytosine.
And it was in the late 90's when mC was recognized as the main cause of epigenetic mechanisms: it is able to switch genes on or off depending on the physiological needs of each tissue.
In recent years, interest in this fifth DNA base has increased by showing that alterations in the methyl-cytosine contribute to the development of many human diseases, including cancer.
Today, an article published in Cell by Manel Esteller, director of the Epigenetics and Cancer Biology Program of the Bellvitge Biomedical Research Institute (IDIBELL), ICREA researcher and Professor of Genetics at the University of Barcelona, describes the possible existence of a sixth DNA base, the methyl-adenine (mA), which also help determine the epigenome and would therefore be key in the life of the cells.
In bacteria and in complex organisms "It was known for years that bacteria, evolutionarily very distant living organisms of us, had mA in its genome with a protective function against the insertion of genetic material from other organisms.
But it was believed that this was a phenomenon of primitive cells and it was very static" describes Manel Esteller.
"However, this issue of CELL publishes three papers suggesting that more complex cells called eukaryotes such as the human body cells, also present the sixth DNA base.
These studies suggest that algae, worms and flies possess mA and it acts to regulate the expression of certain genes, thus constituting a new epigenetic mark.
This work has been possible thanks to the development of analytical methods with high sensitivity because levels of mA in described genomes are low.
In addition it seems that mA would play a specific role in stem cells and early stages of development, "explains the researcher.
"Now the challenge we face is to confirm this data and find out whether mammals, including humans, we also have this sixth DNA base, and consider what its role is".
Source: IDIBELL-Bellvitge Biomedical Research Institute [May 11, 2015]
Classicamente, le basi sono quattro (adenina, citosina, guanina e timina).
Altre due basi derivate da queste sono la metil citosina e la metil adenina, che sono variazioni delle prime due.
Quando si scoprirono le attività biomolecolari importanti delle metil citosina (mC) essa fu aggiunta come quinta base.
Lo studio attuale, di Manuel esteller dell'Università di Barcellona e appena pubblicato sulla rivista Cell, propone che anche la metil Adenina (mA) sia 'promossa' a base; sarebbe quindi la sesta.
Essa è presente in minime quantità in vari batteri (in cui protegge dall'immissione esterna di materiale nucleare) ed in vari vegetali ed animali inferiori (alghe, vermi, mosche) e la sua funzione sarebbe quella di modulare l'attività di vari geni e di influenzare le cellule staminali.
Tutto sta a vedere se si troverà anche nei mammiferi e - in special modo - nell'uomo.
Sixth DNA's base discovered?
DNA (deoxyribonucleic acid) is the main component of our genetic material. It is formed by combining four parts: A, C, G and T (adenine, cytosine, guanine and thymine), called bases of DNA combine in thousands of possible sequences to provide the genetic variability that enables the wealth of aspects and functions of living beings.

Methyl-Adenine 3D structure [Credit: IDIBELL]
Two more bases: the Methyl- cytosine and Methyl-adenine
In the early 80s, to these four "classic" bases of DNA was added a fifth: the methyl-cytosine (mC) derived from cytosine.
And it was in the late 90's when mC was recognized as the main cause of epigenetic mechanisms: it is able to switch genes on or off depending on the physiological needs of each tissue.
In recent years, interest in this fifth DNA base has increased by showing that alterations in the methyl-cytosine contribute to the development of many human diseases, including cancer.
Today, an article published in Cell by Manel Esteller, director of the Epigenetics and Cancer Biology Program of the Bellvitge Biomedical Research Institute (IDIBELL), ICREA researcher and Professor of Genetics at the University of Barcelona, describes the possible existence of a sixth DNA base, the methyl-adenine (mA), which also help determine the epigenome and would therefore be key in the life of the cells.
In bacteria and in complex organisms "It was known for years that bacteria, evolutionarily very distant living organisms of us, had mA in its genome with a protective function against the insertion of genetic material from other organisms.
But it was believed that this was a phenomenon of primitive cells and it was very static" describes Manel Esteller.
"However, this issue of CELL publishes three papers suggesting that more complex cells called eukaryotes such as the human body cells, also present the sixth DNA base.
These studies suggest that algae, worms and flies possess mA and it acts to regulate the expression of certain genes, thus constituting a new epigenetic mark.
This work has been possible thanks to the development of analytical methods with high sensitivity because levels of mA in described genomes are low.
In addition it seems that mA would play a specific role in stem cells and early stages of development, "explains the researcher.
"Now the challenge we face is to confirm this data and find out whether mammals, including humans, we also have this sixth DNA base, and consider what its role is".
Source: IDIBELL-Bellvitge Biomedical Research Institute [May 11, 2015]
domenica 10 maggio 2015
5 - Cercando nel ciarpame.
21.500 geni, si diceva (anche se, per brevità, si cita quel
20.000 che è una cifra tonda più facile da ricordare). Tre miliardi di coppie
di basi, di cui effettivamente usate solo il 2%...

Ragioniamoci su…
Dal punto di vista pratico, sembra uno spreco piuttosto
dispendioso… Se il DNA ‘ciarpame’ fosse davvero inutile, probabilmente non
sarebbe sfuggito alle ‘attenzioni’ di quella che definiamo ‘selezione
naturale’.
Perché?
Perché copiare il DNA richiede energia.
Gli individui che fossero riusciti ad eliminare materiale
genetico inerte, portandosi dentro genomi più parsimoniosi e maneggevoli,
avrebbero quindi avuto un vantaggio biologico reale su quelli che non ci
riuscivano.
Ed un vantaggio biologico di quell’entità significa sicura affermazione.
Ed un vantaggio biologico di quell’entità significa sicura affermazione.
Ma questo evento non si è realizzato, a conti fatti: per
differenza, siamo autorizzati a pensare che esista qualche ruolo ben preciso
per l’EX Dna ‘ciarpame’.
Ed ecco perché il termine non è più in uso…
Il Progetto Genoma Umano riuscì a trovare un bassissimo numero
di geni codificanti proteine: alla fine risultarono essere molto meno dei
100.000 inizialmente teorizzati. Un conteggio certamente troppo basso, inadatto a spiegare tutte le differenze
esistenti tra l’uomo e tutti gli altri organismi. Forse, questo significava che
il genoma ammonta a qualche cosa di più della semplice somma dei suoi geni…
Un esempio tratto dalla vita comune potrebbe essere ottenuto
paragonando un grosso camion ed una piccola macchina sportiva: benché il primo
sia molto più voluminoso, la seconda è infinitamente più agile e veloce.

Rovistare nel ‘ciarpame’.
Si cominciò – pertanto – a guardare con molta più attenzione
quella porzione del genoma che sembrava inutile.
Oggi si pensa che una notevole porzione di esso sia di
origine virale.
Si sa che i virus – per potersi riprodurre nel nostro
organismo – iniettano il proprio genoma all’interno del nucleo cellulare
dell’ospite e così ottengono di fare lavorare le cellule ospitanti per sé,
riproducendosi a spese dell’organismo che hanno infettato.
Si valuta che questi retrovirus endogeni umani costituiscano
oggi circa l’ 8% del totale (pertanto nel ‘libro’ del genoma umano occupano uno
spazio maggiore).
Esistono sequenze virali di varia lunghezza: grandi, medie e
piccole. Alcune furono notate in passato (ripetizioni in tandem) e furono
sfruttate per la cosiddetta ‘impronta genetica’ (vedi il post: “Il Ciarlatano premiato con il Nobel”).
Interessante (e non del tutto infondata, anche se un po’
inquietante) la tesi controcorrente per cui il genoma sarebbe ‘egoista’ e si
riprodurrebbe unicamente per riprodurre sé stesso, a prescindere dalla propria utilità per l’organismo nel quale si
trova (è la tesi di Richard Dawkins, zoologo dell’Università di
Oxford, espressa nel libro “Il Gene Egoista”, 1976).
Un’altra ipotesi è che il DNA non codificante possieda
qualche funzione protettiva sull’integrità, o regolativa sull’attività dei geni
codificanti. Non sappiamo bene quale, né quali meccanismi siano impiegati
eventualmente.
ENCODE: Il consorzio “Enciclopedia od DNA Elements” ha
esaminato dal 2007 circa 30 milioni di coppie di basi (1% del totale!), nel tentativo
di stilare l’elenco dei compiti del DNA. Ciò che ha scoperto fin qui è già
piuttosto significativo. Solo il 2% del genoma è fatto di geni; almeno il 9% è
trascritto su RNA: questo indicherebbe che molto di più del 2% è biologicamente
attivo.
Solo una minima parte di questo RNA è quello detto ‘messaggero’
che porta l’informazione per codificare le proteine.
Il DNA ‘spazzatura’genera vari tipi di DNA: si tratta di
molecole che modificano l’espressione di geni e proteine, con il fine ultimo di
regolare il metabolismo.
A più tardi, Pasuco: sei un paziente ascoltatore, come
sempre…
Iscriviti a:
Post (Atom)