Uso del DNA per
interpretare i cambiamenti di una popolazione.
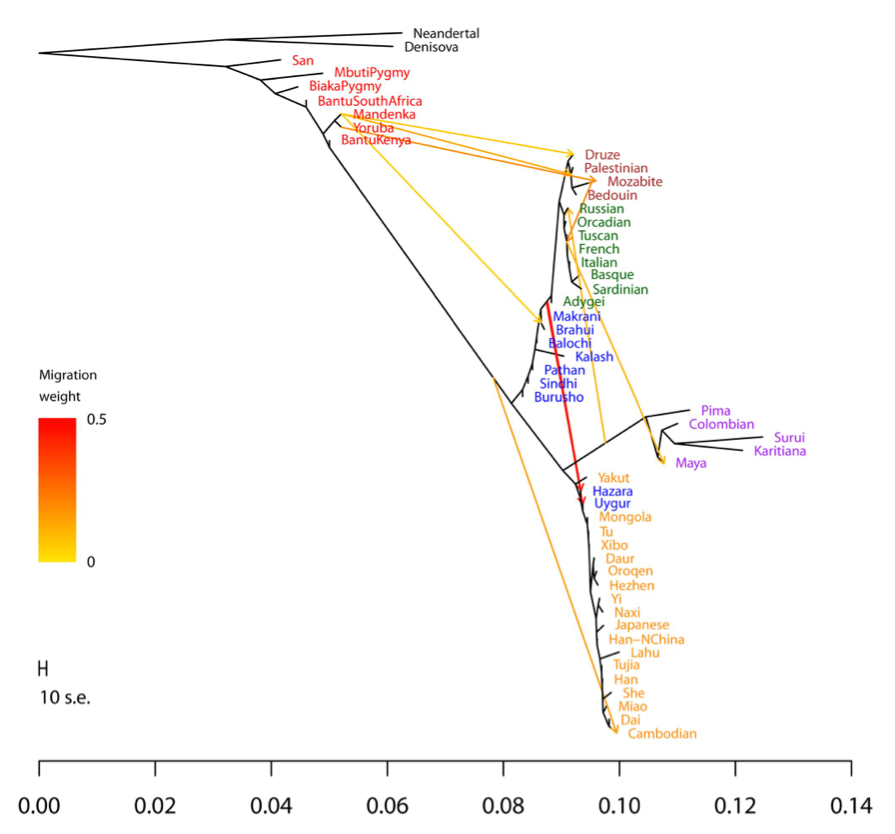 |
| Grafico ad 'albero misto' (Mix Tree, vedi testo) che mostra le previste separazioni e mescolanze tra popolazioni umane studiate [Credit: University of Chicago Medical Center] |
Quando Charles Darwin illustrò per la prima volta il
modo in cui le specie si svilupparono secondo la Selezione Naturale, egli
disegnò quello che sembrava un albero. Il diagramma iniziava al centro, con un
antenato comune, quindi le varie linee si allontanavano una dall’altra, con la
progressiva evoluzione in specie differenti degli organismi.
Nei 175 anni successivi, gli scienziati sono giunti alla
conclusione che il grafico di Darwin sia un po’ semplicistico, dal momento che
le varie specie interagirono tra
di loro in modi che egli allora non considerava neppure possibili (Ma non si
può negare che egli abbia comunque formulato la più importante teoria
scientifica di tutti i tempi, seppure con alcuni dettagli imprecisi). L’uso di un diagramma ad albero è un
modo splendido per illustrare la storia dell’evoluzione di una specie, o la sua
filogenia. Ma non è altrettanto buono per illustrare la storia di popolazioni
di gruppi all’interno di singole specie – come per esempio la specie umana –
che possono spostarsi anche molto e quindi riprodursi tra di loro.
Jonathan Pritchard, professore nel Dipartimento di
Genetica Umana, studia la natura di queste variazioni genetiche combinando
della biologia evoluzionaria e della statistica. Attirato dalle recente
ricerche sul genoma dell’ Uomo di Neanderthal (che suggerisce un maggiore
interbreeding con Homo Sapiens di quanto precedentemente creduto),
Pritchard ha tentato di sviluppare
un metodo generale per valutare il flusso genetico tra gruppi differenti della
stessa specie nel corso del tempo.
In un recente lavoro, pubblicato su PLOS Genetics, insieme a Joseph Pickrell, ricercatore
della Harvard University, ha descritto un modello di software che può definire
l’andamento delle separazioni e
degli incroci fra gruppi all’interno di una medesima specie basandosi sul
moderno DNA.
“ Quando si cerca di ricostruire un diagramma storico ad albero all’interno
di una popolazione, esiste sempre la possibilità di flussi genici da un ramo
all’altro” – afferma Pritchard – “le popolazioni possono incrociarsi se solo
sono geograficamente insieme nel medesimo sito, oppure se c’è movimento da un
sito all’altro. Perciò, la rappresentazione con un albero semplice può non
essere quella più corretta”. Lo scopo della sua ricerca è stato appunto quello
di scoprire quanto di questo schema di albero (lui la
chiama ‘alberosità’, in Inglese) potesse essere conservato e quanto andasse
modificato.
Pritchard and Pickrell hanno sviluppato un software
chiamato TreeMix che confronta
quanto spesso le varianti di uno stesso particolare gene proveniente da
differenti popolazioni compare
nella medesima specie. Quindi,
calcola quanto questi gruppi siano strettamente imparentati e quando nella loro
storia essi si siano separati per formare una precisa popolazione separata e
distinta.
Il grafico risultante possiede ancora un aspetto in
qualche modo vegetale, ma si allontana un poco dalle nette ramificazioni
arboree per assomigliare più ad un intricato cespuglio, o alle ramificazioni di
una vite. Il tronco del cespuglio rappresenta la principale e maggiore
relazione tra i gruppi, ed i rami più grandi rappresentano le poplazioni distinte
che si sviluppano man mano nel tempo, da sinistra a destra nel grafico. Ma il
dettaglio più ragguardevole sono gli intrecci tra i rami, che mostrano gli
eventi migratori a mezzo dei quali una popolazione precedentemente separata si
è mescolata con un’altra, ricompattandosi in seguito per formare un nuovo
gruppo distinto.
Pritchard and Pickrell hanno provato il proprio modello
usando il DNA di 55 popolazioni e di 82 ‘razze’ canine. Ed hanno già trovato
alcuni risultati interessanti.
Per esempio: il DNA dei cani boxer (9%) e dei
basenji (25%) può essere
rapportato al lupo anche dopo il loro addomesticamento, il che vuol dire che questi gruppi hanno fatto ricorso
ad interbreeding anche dopo che l’addomesticamento umano era inziato.
Insomma: questo nuovo metodo permette di individuare e rappresentare interrelazioni più complicate di quanto
non permetta il vecchio sistema ad albero ‘tradizionale’.
Un altro esempio: i Mozabiti algerini possiedono un DNA che è largamente un
miscuglio di eredità europeo e medio orientale. Ma si sono anche mescolati in vari momenti della loro
storia con antenati di popolazioni sub-sahariane. Il nuovo modello misto può
rappresentare questi complessi andamenti, che il vecchio schema ad albero
descriveva solo con “antenati prevalentemente medio orientali”.
Un altro gruppo di ricercatori ha già utilizzato questo
software per dimostrare l’esistenza di un collegamento tra i Denisoviani
(parente estinto del Neanderthal in Siberia) ed i Pappasi del Sud Pacifico. Si
tratta di un dato che non ha un senso immediato – almeno geograficamente – ma che
costringerà i ricercatori a porsi nuove domande circa le migrazioni nel tempo
dei rispettivi gruppi interessati.
C’è da attendersi molti chiarimenti, con questo metodo,
specialmente al riguardo di quelle latitudini nelle quali si sono avuti, nel
corso del tempo, ripetute e confuse migrazioni d’intere popolazioni, come ad
esempio l’Anatolia. E qualche correzione della Storia.
Author: Matt Wood | Source: University of Chicago Medical Center
[January 08, 2013]
Diffida: il presente testo ed i grafici non possono essere superficialmente riportati, citati o interpretati da soggetti non addetti ai lavori, né strumentalizzati per fini politico-identitari da minoranze linguistiche.
Diffida: il presente testo ed i grafici non possono essere superficialmente riportati, citati o interpretati da soggetti non addetti ai lavori, né strumentalizzati per fini politico-identitari da minoranze linguistiche.